

Living with Sickle Cell Anemia
By Remi Agbabiaka
I got a watspp message from a very close friend during the Sickle cell Awareness Week, and it reminded me of that very silent killer disease. Despite its death rate, people have continued to defy it and its effects. Whilst lots of attention need be paid to researches in the treatment of the disease, prevention is the best way out for the world. Whilst the medical world continues to seek cures for the disease however, it is also important encourage, love and embrace the sufferers of the disease rather than stigmatize them and that’s the crux of the message.
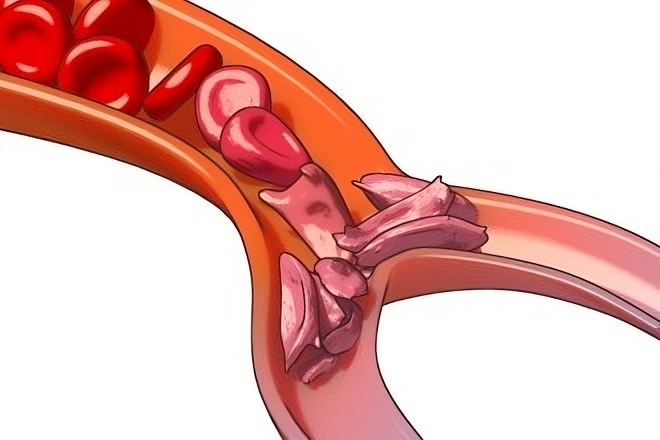
Sickle-cell anemia, also called sickle-cell disease, is a hereditary disorder in which abnormal haemoglobin within the red blood cells (RBCs) causes the cells to take on abnormal sickle (crescent) shapes. This decreases the ability of the hemoglobin to transport oxygen throughout the body. The sickled cells tend to bunch up and clog the blood vessels causing inflammation, pain, tissue damage, and anemia. Sickle cell anemia is caused by a mutation in the gene that tells your body to make hemoglobin — the red, iron-rich compound that gives blood its red color. Hemoglobin is a component of every red blood cell. It allows red blood cells to carry oxygen from your lungs to all parts of your body, and to carry carbon dioxide waste from throughout your body to your lungs so that it can be exhaled. Under normal circumstances, the body makes healthy hemoglobin known as hemoglobin A. People with sickle cell anemia make hemoglobin S — the S stands for sickle. Sickle cell disease is a genetic disorder, its frequency varies in different populations worldwide. It is found most frequently in Africa, where in some locations up to 40 percent of the population has at least one HbS gene. The gene is also found in people in Mediterranean and Middle Eastern countries, such as Italy, Greece, Saudi Arabia, India, Latin America, the Caribbean, and the United States. Because sickle-cell disease is hereditary, people are born with it and will have it all their lives. There is no way one person can catch sickle-cell disease or sickle-cell trait from another, and there is no way the disease can be cured.
Manifestation

Sickle cell is manifested in two forms, the Sickle Cell trait and the Sickle cell disease. People inherit one set of genes from each parent. They can inherit either two normal hemoglobin genes (HbA), one normal hemoglobin gene and one gene for the abnormal hemoglobin gene (HbS), or two abnormal HbS genes, depending on the composition of their parents’ genes.
When a person carries one HbS and one HbA gene, the presence of the normal gene is sufficient to override the effects of the HbS gene, so the symptoms of sickle-cell disease do not develop. These people, however, are said to have the sickle-cell trait or to be carriers of the sickle-cell trait, and they may pass it on to their children. Prospective parents who are likely to carry the HbS gene may wish to be tested for its presence and receive genetic counseling before having children.
When people inherit two abnormal HbS genes, one from each parent, they have sickle-cell disease, and they show symptoms of that disease. They have only abnormal HbS genes to pass on to their children.
Complications
People who have sickle-cell disease get infections more frequently than other people. Babies with sickle-cell disease are often given antibiotics to prevent infections. Before this screening became common, many babies born with sickle-cell disease died in infancy. Today the use of preventive antibiotics has significantly reduced the number of babies who die. Infants, older children, and adults with sickle-cell disease also periodically experience bouts of critical illness called crises. They also suffer from complications of anemia.
Crises:Sickle-shaped red blood cells clump more easily than normal RBCs. Sickle-cell crises start suddenly when clumping of sickled RBCs in the blood vessels obstructs the normal flow of blood, depriving various tissues and organs of oxygen. The first crises usually appear in early childhood. Crises may be brought on by respiratory infection, by a loss of body fluids from vomiting or diarrhea, by situations where the body’s need for oxygen is increased, or they may occur for no obvious reason. They may last for several days, and cause fever and sharp, intense pain in the back, abdomen, chest, arms and legs. In infants, the hands and feet may become swollen and painful.
Crises may damage nearly any part of the body, but especially the bones, kidneys, intestines, lungs, liver, spleen, and the central nervous system *, including the brain. There may also be eye damage, stroke, convulsions, or paralysis. The damage is caused because the clumping of RBCs in a blood vessel deprives tissues of oxygen. Many people with sickle-cell disease go for long periods during which they may feel relatively well and engage in most normal activities and are free of crises (in remission). Others may experience pain on a daily basis, and some need to be hospitalized as a result of crises several times a year.
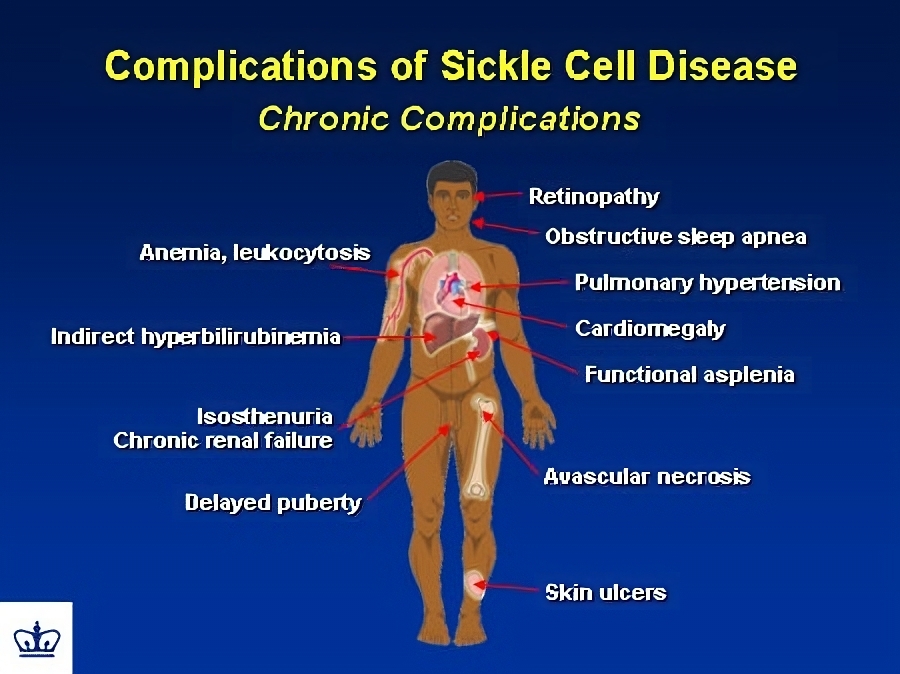
Anemia:Sickle cells are more easily broken down and destroyed than normal RBCs. Sickle cells have a lifespan of 10 to 20 days compared to 90 to 120 days for a normal RBC. People with sickle-cell anemia thus cannot keep up a normal level of oxygen-carrying hemoglobin in their blood. This is so despite the fact that they make red blood cells faster than people without the disease. The result is that they are anemic. The anemia may become so severe that a person will need to have a blood transfusion. The long-term effect of anemia is that the heart has to work harder to pump more blood through the body. Over time the heart enlarges, increasing the risk of heart attack and heart failure.
Diagnosis
Sickle-cell disease and sickle-cell trait can be diagnosed by a test that is much simpler than its name: hemoglobin electrophoresis. This test detects the presence of HbS and other abnormal hemoglobins.
Treatment
There is no cure for sickle-cell anemia. Much treatment is preventive and directed toward symptoms. Antibiotics may be given to prevent infections. Fluid intake is important to prevent dehydration which is a major cause of sickling. Folic acid may be given daily to lessen the anemia by helping to make new red cells. Children are given a complete set of immunizations. Lifestyle habits that can help sickle-cell patients stay healthy and have fewer crises include drinking plenty of water, avoiding extremes of heat and cold, avoiding stress and overexertion, getting enough sleep, and having regular medical check-ups.
Treatment in a sickle-cell crisis may require oxygen therapy, pain relieving medications, antibiotics, and intravenous fluids to offset dehydration. Blood transfusions may also have to be performed. Treatment of pain is a major concern for physicians whose patients have sickle-cell disease. The benefits of different pain relievers and their unwanted side effects must be balanced for each patient.
Research is ongoing to find better ways to treat people with sickle-cell disease. Some of these research efforts are directed at stimulating the production of fetal hemoglobin, a form of hemoglobin found in infants, even those with sickle-cell disease. Other research is directed toward the development of drugs that block dehydration in cells and the transplantation of healthy bone marrow that makes normal red blood cells are also under investigation.
Risk factors
The risk of inheriting sickle cell anemia comes down to genetics. For a baby to be born with sickle cell anemia, both parents must carry a sickle cell gene. It is advised that every man or woman go for blood test to ascertain their blood genotype. The reduction or eradication of this disease can only be through the avoidance of genotypically incompatible adults refusing to get married to prevent having children with the disease.
Its not all gloom though, the individual with the sickle cell trait (Hb AS) is protected from sickle cell symptoms and complications by the presence as well of Hb A. He is largely protected from death caused by malaria, especially in early childhood, by the presence of Hb S. Thus he enjoys the best of both worlds and is naturally better adapted to the malaria infested environment than the individual with the normal Hb AA who is more liable to childhood death from malaria, or the sickle cell affected one with Hb SS in whom malaria easily induces a lethal degree of anaemia.
Those with sickle cell anemia can lead productive, full lives with good medical care. The life expectancy for those with sickle cell anemia has increased due to better treatment and healthcare options; those with the condition now live into middle age (forties and fifties), and older. For those living with sickle cell anemia, the following are key points to remember in order to live a healthy life, Adopt or maintain a healthy lifestyle, Take steps to prevent and control complications, Learn ways to cope with pain.














